
| мҙҲлЎқ | Home > мҙҲлЎқ |
мҙҲмІӯк°•м—° 1
к°•мӨҖкө¬ (лҢҖкө¬кІҪл¶Ғкіјн•ҷкё°мҲ мӣҗ)
-Title: General gauge symmetry in the theory and simulation of atomic heat transport
-Abstract: The Green-Kubo theory of atomic heat transport relies on the ambiguous decomposition of total energy into ‘atomic’ energies of individual atoms. The challenge is to understand how the transport coefficient thermodynamically emerges from such ill-defined atomic energies. Here, we show that a simple symmetry principle for atomic energies (that is, “all possible ways of distributing energy among atoms are equivalent”) dictates the general theory of atomic heat transport. To this end, we define atomic gauges that regulate the redundant degrees of freedom in atomic energies. The atomic gauge symmetry then uniquely determines the gauge-invariant form of macroscopic energy transfer, which is identified as heat. Consequently, arbitrary choices (even random shuffling) of atomic energies lead to the same heat conductivity k. The gauge theory not only lays a firm foundation of the Green-Kubo formalism of heat transport, but also offers a novel variational method for calculating k of non-solid materials, as numerically demonstrated using machine learning-inferred atomic energies of Cu2S.
мҙҲмІӯк°•м—° 2
л°•мІ нҷҳ (м„ңмҡёлҢҖн•ҷкөҗ)
-Title: Impurity-Strength Dependent Quasiparticle Interference and Gaussian Time-Dependent Variational Principles for Anharmonic Lattices
-Abstract: In this talk, I will discuss some of the recent projects of my research group. First, I will show that the common treatment of the Fourier-transformed scanning tunneling spectroscopy of a material with impurities by using the joint density of states, or by the Born approximation, both of which are devoid of the information of the impurities, can break down severely in simple materials such as a square lattice with an s-like orbital per site as well as complex ones such as TaAs. Then, I will show that the dynamical ansatz for the self-consistent harmonic approximation for anharmonic lattices can be proven using the time-dependent variational principles within the Gaussian manifold and suggest promising research directions based on our findings.
References
[1] S.-J. Hong, J.-M. Lihm, and C.-H. Park, J. Phys. Chem. C 125, 7488 (2021).
[2] J.-M. Lihm and C.-H. Park, arXiv:2010.15725.
мҙҲмІӯк°•м—° 3
мқҙмһ¬лҸҷ (лҢҖкө¬кІҪл¶Ғкіјн•ҷкё°мҲ мӣҗ)
-Title: Ultrafast dynamics of Dirac semiconductors
-Abstract:Ultrafast dynamics in the Floquet states of Dirac semiconductors are explored, which accentuates nonequilibrium dynamical nature of the Floquet state beyond the usually considered quasi-static band nature and then gives a missing connection between the two. Phase oscillation of the Fano resonance at the frequency of 2ω_pump on the Floquet Dirac cone of graphene is disclosed from the transient absorption spectroscopy (TAS), which is due to the correlation between 2ω_pump-absorption paths. This establishes a driving of the petahertz (PHz) quantum oscillation from Dirac materials. On another Floquet Dirac cone of the graphene/h-BN (Gr/h-BN) heterostructure under the circularly polarized pulse pumping, the fluctuating topological order (FTO) is discovered to exist between topologically trivial and optically induced nontrivial phases from the time-resolved dichroic photoemission spectroscopy (TRdPES). The topological order fluctuation defines a novel mixed phase, i.e., a dynamical mixture of trivial and nontrivial phases, as a precursor to the transient nontrivial phase.
мҙҲмІӯк°•м—° 4
к°•м°Ҫмў… (충лӮЁлҢҖн•ҷкөҗ)
-Title: Nature of electronic correlations in a strongly correlated system: the infinite-layer nickelate
-Abstract: Strongly correlated systems show interesting physical phenomena including superconductivity, magnetism, and metal-insulator transitions. In order to study the origin and nature of electronic correlations in a strongly correlated system, first principle calculations including density-functional theory (DFT) and DFT plus dynamical mean-field theory (DFT+DMFT) were performed. In the infinite-layer nickelate, the optical conductivity calculations enable us to identify the origin and nature of electronic correlations. Besides, we found a crossover from Mott to Hund region in the nickelate systems, where Hund’s physics is hidden at low energies but noticeable at intermediate energies. This kind of hidden Hund’s physics is quite new and thus opening a new research area in the theory of correlated systems.
In the second topic of this talk, we will discuss the methodology of computational correlated material design. The design of correlated materials challenges researchers to combine the maturing, high throughput framework of DFT-based materials design with the rapidly-developing first-principles theory for correlated electron systems. We introduce a material design workflow, and illustrate it via some examples, highlighting the interplay between theory and experiment with a view towards finding new materials. We review the statistical formulation of the errors of currently available methods to estimate formation energies. We formulate an approach for estimating a lower-bound for the probability of a new compound to form. Correlation effects have to be considered in all the material design steps. These include bridging between structure and property, obtaining the correct structure and predicting material stability. We introduce a post-processing strategy to take them into account. Finally, we discuss future directions for computational material designs.
мҙҲмІӯк°•м—° 5
мӢ¬м§ҖнӣҲ (нҸ¬н•ӯкіөкіјлҢҖн•ҷкөҗ)
-Title: The role of Hund's coupling on approach to metal insulator transition in NiS2−xSex
-Abstract: Understanding the characteristic energy scale is a fundamental issue in a strongly correlated electron system. In the multiband system, the energy scale is not only affected by the effective Coulomb interaction but also by the Hund’s coupling. In an angle resolved photoemission spectroscopy of NiS2-xSex system the evolution of kink has been observed, and a dynamical mean field theory calculation combined with density functional theory confirm that this kink is originated from Hund’s coupling.[1,2] The abrupt deviation from the Fermi liquid behavior in the electron self-energy makes kink features at low energy scale and this kink is directly related to the coherence-incoherence crossover temperature scale. The evolution of the characteristic temperature scale via kink features in the spectral function is the hallmark of Hund’s physics in the multiorbital system.
[1] C.-Y. Moon et al. Physical Review B 92, 235130 (2015).
[2] B.G. Jang et al. Nature Communications 12, 1208 (2021).
мҙҲмІӯк°•м—° 6
мқҙмҳҲлҰ¬ (н•ңкөӯнҷ”н•ҷм—°кө¬мӣҗ)
-Title: Data-driven research using machine-learning approach for materials design
-Abstract: Data-driven research using machine-learning (ML) approaches has been applied extensively in a variety of daily life, industry, and research fields from a completely new perspective. In scientific research as well, various attempts have been made to apply ML approaches to problems that require very high costs, or problems that are too complex to be solved within conventional methods. The recent development of qualified open-access materials databases has also accelerated data-driven studies in physics, chemistry, and materials research, such as Materials project, Open quantum materials database (OQMD), Automatic-Flow for materials discovery (AFLOW), and so on. In this presentation, I will introduce an example study on the materials design of highly efficient thermoelectric materials, especially on SnSe-based alloys, through data-driven research using the ML method. The thermoelectric effect is one of the core technologies for recycling energy by converting heat to electricity. It has been the ultimate goal to achieve high-performance thermoelectric materials through optimizing materials. In this study, we performed a full-cycle of data-driven research to find optimization principles and knowledge for high-efficiency thermoelectric materials. It consists of data generation of experiments and computations, building a ML predictive model, optimizing the thermoelectric materials, and its applications. This ML-based predictive model allows fast screening of high-dimensional material space, and suggests six best SnSe-based alloying compositions with high thermoelectric performance. Our research is expected to provide new high-efficiency thermoelectric material candidates as well as new research strategies for materials design through a data-driven research.
мҙҲмІӯк°•м—° 7
мқҙмқёнҳё (н•ңкөӯн‘ңмӨҖкіјн•ҷм—°кө¬мӣҗ)
-Title: Crystal structure prediction in a representation space
-Abstract: In this talk I present a method of finding multiple crystal structures similar to the known crystal structures on database through variational autoencoder and Pearson’s coefficient. The radial distribution function (RDF) is used to represent the known crystal structures, and then the variational autoencoder is employed to generate a set of replica RDFs. For given chemical compositions and crystal volume, we generate random crystal structures using constraints for space group and Wyckoff positions and directly compare their RDFs with those of the known and/or replicated RDFs using Pearson’s coefficient. For selected crystal structures, energy minimization is subsequently performed through first-principles calculations. This approach enables us to predict a set of new low-energy crystal structures using only the information on the RDFs of the known structures.
мҙҲмІӯк°•м—° 8
мқҙмғҒмҡұ (н•ңм–‘лҢҖн•ҷкөҗ)
-Title: Designing Descriptor for Computational Screening of Argyrodite-based Solid State Superionic Conductors
-Abstract: All-solid-state rechargeable lithium-ion batteries are attractive power sources for electrochemical applications due to their potential to improve safety and stability compared to conventional batteries with liquid electrolytes. Finding a solid electrolyte with high ionic conductivity and compatibility with other battery components is a key factor in increasing the performance of any solid LIB. Several structural and chemical families of solid LIB conductors such as LGPS, LISICON and Argyrodites have been shown to exhibit ionic conductivity spanning over 10mS/cm at room temperature (RT). And Lithium argyrodites make up a promising family of solid state electrolytes, characterized by the general composition of Li7-y-x[(A1-y/By)C4]C1-xX1+x (A, B, C and X denote elements of group IV, V, VI and VII, respectively) Here, we present systematic exploration of descriptors for computational screening of the argyrodite-based solid state superionic conductors. Argyrodite consist of polyanions and single anions surrounded by cage-like Li ion diffusion network. We found that the ionic conductivity has strong correlation with the disorderedness of single anions and the size of the Li-ion cage formed around the single anions. Especially, when the Li-ion cages are of uniform size, the inter-cage diffusion for higher ionic conductivity is accelerated. Therefore, we proposed the uniformity of Li-ion cage size as a screening descriptor for argyrodite-based solid state superionic conductors. The proposed our results will provide insights into the development of new compositions of argyrodite. In addition, we found that halogens play an important role in inter-cage diffusion because Li-ions prefer to reside around the halogen ions in small numbers. Our results will provide new approaches in the compositional changes in Li argyrodite to increase Li-ion conductivity based on the atomistic understanding of origin of inter-cage diffusion.
мҙҲмІӯк°•м—° 9
Stefan Ringe (лҢҖкө¬кІҪл¶Ғкіјн•ҷкё°мҲ мӣҗ)
-Title: Computational catalyst design at electrified solid-liquid interfaces
-Abstract: Electrochemistry is at the heart of a future sustainable energy landscape, driving the green conversion and storage of energy. To date, however, almost all electrochemical processes suffer from a high overpotential or low product selectivity which greatly reduces their industrial competitiveness. In the search for more active and selective electrocatalysts, quantum chemical simulations based on Density Functional Theory have become a state-of-the-art means. In order to computationally facilitate the involved complex calculations, the electrochemical interface is commonly highly simplified by ignoring physical characteristics such as the accumulation of surface charge leading to the creation of the double layer, solvation stabilization of reaction intermediates or mass transport effects. In this talk, we first present continuum solvation methods as a way to to beyond these approximations and then use them to revisit such effects with a particular focus on electrochemical CO2 reduction. In contrast to previous believes, we find the interfacial electric field to be significant and even determine the product selectivity and conversion rate on Gold and Copper catalysts. We show how this observation leads to specific design principles for the electrolyte and catalyst for which we perform a descriptor-based screening study and identify promising catalysts. The idea is extended to other electrochemical systems, for which we find solvation effects to be a further critical factor. Finally, by introducing a multi-scale computational design approach, we show that also mass transport is critically influenced by the electrified interface and plays a crucial role in understanding electrochemical processes.
мҙҲмІӯк°•м—° 10
мқҙмӨҖнқ¬ (мҡёмӮ°кіјн•ҷкё°мҲ мӣҗ)
-Title: Ultimate-density ferroelectric memory via flat phonon bands
-Abstract: While flat energy bands in electrons are known to cause exotic phenomena such as graphene superconductivity, flat bands in phonon are barely investigated. We discover that flat phonon bands exist surprisingly in a commercial ferroelectric HfO2 and that they produce ultimately-localized dipoles within its half-unit-cell-widths, ~3в„« [1]. Irreducibly localized, these dipoles are impervious to any external effects such as domain walls, surface exposure, and even down-to в„«-scale miniaturization. More strikingly, these independently acting dipoles are individually switchable without creating any domain-wall energy cost. With its direct integrability onto Si-technology, flat bands in HfO2 bestow opportunities for storing bits each in its every individual unit-cell toward an ultimately-dense memory device [2].
[1] Lee et al., “Scale-free ferroelectricity induced by flat phonon bands” Science 369, 1343 (2020)
[2] Noheda et al., “A key piece of the ferroelectric hafnia puzzle” Science 369, 1300 (2020) (“Perspective” on our paper, Ref. [1])
мҙҲмІӯк°•м—° 11
м •мһ¬мқј (м„ңмҡёмӢңлҰҪлҢҖн•ҷкөҗ)
-Title: Gate tunable valley polarization and topological moire bands in ABC-trilayer graphene
-Abstract: The enhanced density of states near charge neutrality in ABC trilayer graphene due to the flattening of the band dispersion makes this type of multilayer graphene systems prone to form correlated ordered phases in sufficiently clean devices. We show by means of mean field Hartree-Fock calculations with long-range Coulomb interactions that the energetically favored ground state for ABC trilayer graphene turns from the layer antiferromagetic (LAF) phase to the valley polarized anomalous Hall (AH) phase upon a small carrier doping and application of a perpendicular electric field. Our phase diagram for the different spin-valley flavor polarization indicates that a single flavor polarization giving rise to an anomalous Hall phase is favored for small carrier densities and large displacement fields, while three flavor polarization is generally suppressed. The uneven filling of the three electron or hole pockets near the band edges upon carrier doping makes the system prone to form nematic phases with broken rotational symmetry due to momentum space condensation, an exchange driven mechanism that tends to lower the energies of electronic states that are closer together in momentum space. We further comment on the possibility of generating nearly flat moire bands with quantized valley Chern numbers by introducing a hexagonal boron nitride substrate close to alignment, and show that off-diagonal vector potentials that break the C3 rotational symmetry can trigger topological phase transitions that are in keeping with experimental observations.
мҙҲмІӯк°•м—° 12
진кІҪнҷҳ (кё°мҙҲкіјн•ҷм—°кө¬мӣҗ)
-Title: Electronic and topological properties in 1T-TaS2 flat band material
-Abstract: Effects of electron many-body interactions amplify in an electronic system with a narrow bandwidth opening a way to exotic physics. 1T-tantalum disulfide (1T-TaS2) has spurred considerable interest, due to its flat band and the Mott insulating behavior induced by the charge density wave formation. But the nature of the low-temperature insulating state in 1T-TaS2 has remained unresolved due to the strong correlations, electron-phonon interactions and structural ambiguity. Here, we show the coexistence of two different surface terminations, which exhibit distinct behaviors for the K adsorption in both adsorption sites and in local electronic states. These behaviors were straightforwardly interpreted through theoretical calculations to indicate the distinct electronic properties of the different terminations, that is, a Mott insulating state and a trivial band insulating state. Beyond this adsorption behavior, at 1/3 monolayer coverage, we successfully fabricated ordered superstructures of adsorbates, which leave 2/3 of surface unitcells in a honeycomb lattice. On its Mott insulating phase, this lattice represents a honeycomb Mott insulator with active topological degrees of freedom. This is one of the first material realization of the Kane-Mele-Hubbard Hamiltonian and provides an extremely interesting platform to seek novel emerging phases as discussed in twisted bilayer graphene.
мҙҲмІӯк°•м—° 13
к¶ҢмҳҒк· (кІҪнқ¬лҢҖн•ҷкөҗ)
-Title: Giant Hidden Rashba Effect in Two-dimensional Bi2Si2 with an Inversion Symmetry
-Abstract: In addition to spin-orbit coupling (SOC), it is an asymmetric crystal potential that induces Rashba (R-1) spin splitting observed at surfaces or interfaces. Recently, it has, however, been found that even centrosymmetric materials can exhibit a similar but distinct spin splitting called “hidden” Rashba (R-2) effect manifesting spatially segregated spin splitting or spin-layer locking (SLL) as shown in the figure below. To understand the underlying physical origin of such R-2 phenomena, we used the first-principles density functional theory and model Hamiltonian calculation to investigate a new two-dimensional (2D) materials Si2Bi2, which we identified to possess an ideal condition for the strong R-2 SLL. Our study revealed that the hidden SLL can be determined by a competition between the SOC and sublayer-sublayer interaction. We evaluated the Rashba strength to be 2.16 eVÅ, which is the greatest value ever observed in 2D R-2 material to the best of our knowledge. [1]
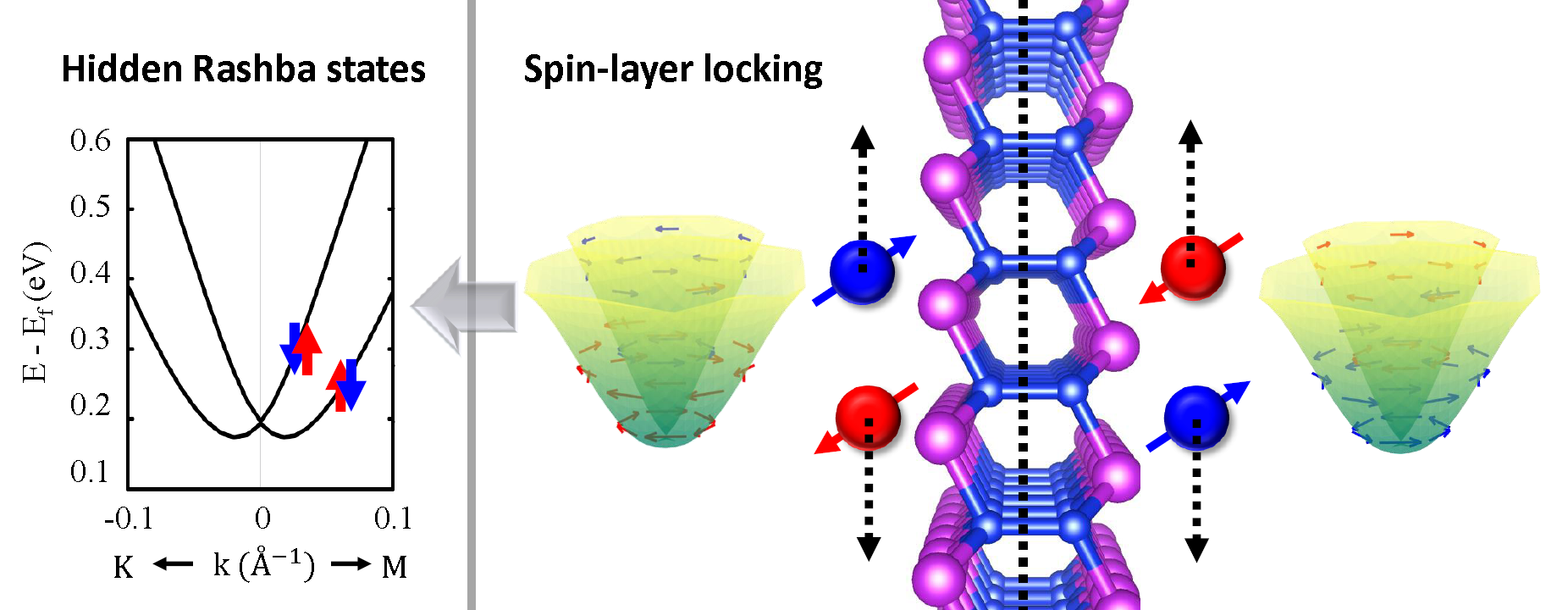
Hidden Rashba states corresponding to the spatially segregated Rashba spin splitting called spin-layer locking observed in an equilibrium phase of Si2Bi2 with the inversion symmetry.
[1] Seungjun Lee and Young-Kyun Kwon, Unveiling Giant Hidden Rashba Effects in Two-Dimensional Si2Bi2, npj 2D Mater. Appl. 4, 45 (2020).